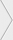使用方法
簡易解析機能
■BWA解析
(1)解析ジョブ一覧の上部にある「新規BWA解析」ボタンをクリックして、「Mapping and Calling SNPs」画面を表示します。
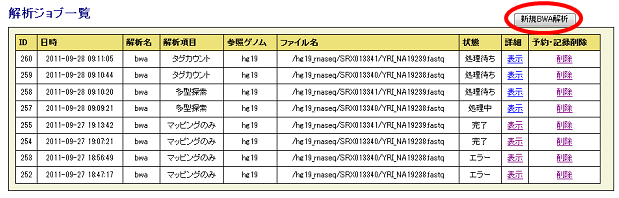
(2)解析項目(マッピングのみ、多型探索、タグカウント)、入力ファイル(Single/Pair、ファイルの選択)、対象ゲノムをそれぞれ選択します。
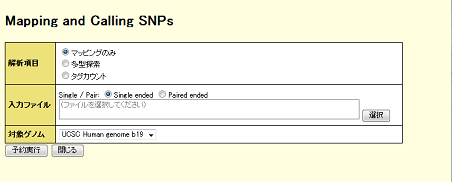
(3)入力ファイルの「選択」をクリックすると、「Select FASTQ file for input」画面が表示されます。画面左側のディレクトリを選択すると、画面右側にディレクトリ内のファイル一覧が表示されます。表示されたファイルの一覧から解析対象のfastqファイル(拡張子がfastq、fqのファイルのみが選択可能です)を選択し、OKボタンで確定します。
※解析に利用するfastq形式ファイルは/genome_box/projectディレクトリ内にあるファイルのみが対象となります。必要に応じて予めコピーしておいてください。
(4)「Mapping and Calling SNPs」画面で「予約実行」ボタンをクリックすると解析ジョブ一覧にジョブが登録されます。予約実行すると入力ファイルと同じディレクトリに新たなディレクトリ(ディレクトリ名は「BWA_YYYY-MM-DDTHH:MM:SS」の形式。各々年月日時分秒が入ります)が作成され、解析結果がそのディレクトリ内に出力されます。出力されるファイルの一覧は下記のようになっています。
○解析項目およびSingle/Pairの設定別出力ファイル
| 解析項目Single/Pair | 出力ファイル | マッピングのみSingle | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.sam |
|---|---|
| マッピングのみPair | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.samBWA_result_inconsistent.samBWA_result_oneside-unmapped.sam |
| 多型探索Single | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.samBWA_result.bamBWA_result.sort.bamBWA_result_raw.bcfBWA_result.vcf |
| 多型探索Pair | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.samBWA_result_inconsistent.samBWA_result_oneside-unmapped.samBWA_result.bamBWA_result.sort.bamBWA_result_raw.bcfBWA_result.vcf |
| タグカウントSingle | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.samBWA_result.bamBWA_result.sort.bamBWA_result.tagcount.bedgraph |
| タグカウントPair | run_log.txtBWA_log.txtBWA_result.samBWA_result_unmapped.samBWA_result_inconsistent.samBWA_result_oneside-unmapped.samBWA_result.bamBWA_result.sort.bamBWA_result.tagcount.bedgraph |
○各出力ファイルについて
| ファイル名 | 内容 |
|---|---|
| run_log.txt | 全解析のログファイル |
| BWA_log.txt | BWAのログファイル |
| BWA_result.sam | マッピングされたリードのみを含むSAMファイル |
| BWA_result_unmapped.sam | マッピングされなかったリードのみを含むSAMファイル |
| BWA_result_inconsistent.sam | Paired endedで同じ方向にマッピングされたリードのみを含むSAMファイル |
| BWA_result_oneside-unmapped.sam | Paired endedで一方のリードがマッピングされなかったリードのみを含むSAMファイル |
| BWA_result.bam | BWA_result.samをBAM変換したファイル |
| BWA_result.sort.bam | BWA_result.bamをソートしたBAMファイル |
| BWA_result_raw.bcf | 多型探索により作成されたbcfファイル |
| BWA_result.vcf | 多型探索により作成されたvcfファイル |
| BWA_result.tagcount.bedgraph | タグカウントにより作成されたbedgraphファイル |